 百木园
百木园1958年F.H.C. 克里克提出了生物学中重要的中心法则,DNA->RNA->蛋白质,中心法则说明,DNA可以转录形成RNA,RNA再翻译成一个个氨基酸,最后组合形成蛋白质。
通过中心法则不难看出,如果把DNA比喻为进行工业生产的设计蓝图,那么蛋白质就像实现这个蓝图的工具,所以说蛋白质是一切生命活动的基础,它几乎参与了所有的生物学过程,如遗传、发育、繁殖等等。对蛋白质进行深入地研究,能让我们从更深层次诠释生命体的构成和运作变化规律,进而全面揭示生命运行、发展的机制,激发生物科学、药物研发、合成生物学、酶科学等领域的发展。
因探究生物体内各种蛋白质的功能及其机制等是目前蛋白质研究的主要内容,同时也是后基因组时代生命科学领域的主要研究热点之一。蛋白质的功能很大程度上取决于蛋白质的结构,因此如何破解蛋白质的三维结构成为了科学家研究的重点。
AlphaFold2的诞生
近些年来,随着人工智能技术的发展,深度学习等相关技术也被应用在蛋白质结构预测领域。2018年的CASP 13(国际权威的蛋白质结构预测竞赛,每2年举办一次)上,谷歌DeepMind团队的AlphaFold拿下了70多分,打败众多研究团队,取得人工组第一,在该领域取得了里程碑式的进展。在2020年的CASP 14上,谷歌DeepMind团队的AlphaFold2以惊人的92.4分登顶第一[1],这一结果也被认为是基本解决了“困扰了生物学家50年”的问题,获得重大突破。92.4分,指的是对竞赛目标蛋白的预测精度GDT_TS分数达到92.4,一般认为该分数超过90分,基本可以替代实验方式啦,这也意味着AlphaFold2预测的结果与实验得到的蛋白质结构基本一致。
2021年7月15日, DeepMind团队在国际顶级期刊《Nature》上发表论文,详细描述了AlphaFold2的设计思路,并提供了可供运行的基于JAX的模型和代码[2]。考虑到JAX受众偏向专业的AI科学计算研究人员,且飞桨社区尚没有蛋白质结构预测相关的开源项目,百度螺旋桨PaddleHelix生物计算团队,基于飞桨深度学习框架,复现了AlphaFold2模型,提供给广大飞桨开发者使用,帮助大家快速入门蛋白质结构预测。
https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/paddlefold
AlphaFold2算法的设计思路
AlphaFold2通过独特的神经网络和训练过程设计,第一次端到端地学习蛋白质结构。整个算法框架通过协同学习蛋白质的多序列比对(MSA)和氨基酸对(pairwise)的表征,将蛋白质序列的进化信息、蛋白质结构的物理和几何约束信息结合到深度学习网络中。我们将从数据预处理、Evoformer和Structure Module三个模块分析AlphaFold2算法的设计思想。
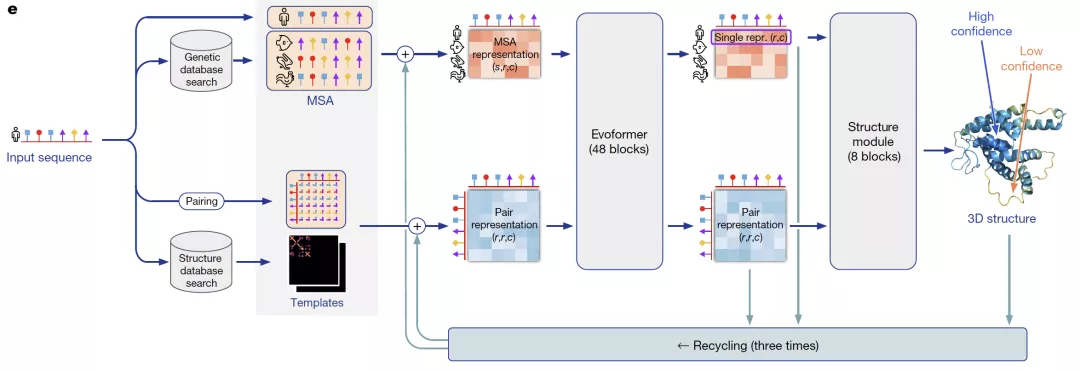
来自:AlphaFold2论文
数据处理
预测蛋白结构时,AlphaFold2会利用氨基酸序列信息在蛋白质库中搜索多序列比对(MSA)。MSA可以反映氨基酸序列中的保守性区域(即不容易产生突变),这些保守性区域和蛋白质的结构息息相关,比如可能被折叠在蛋白质内层,不容易和外界产生相互作用,进而不易受影响发生突变。在AlphaFold2的数据预处理中,为了减少模型运算量,会先对MSA中的序列进行聚类,取每个类别中心的序列作为main MSA特征。除了MSA,AlphaFold2的另一个重要输入是氨基酸对(pairwise)的特征。作为main MSA的补充,Alphafold2会随机采样非聚类中心的序列作为extra MSA输入一个4层的网络提取pairwise特征,然后和模版提取的pairwise特征相加后得到最终pairwise特征。main MSA特征和pairwise特征通过48层Evoformer进行表征融合。
Evoformer
Evoformer网络的设计动机是想利用Self-Attention机制学习蛋白质的三角几何约束信息,同时让MSA表征带来的共进化信息和pairwise表征的结构约束信息相互影响,使得模型能直接推理出空间信息和进化信息的联系。
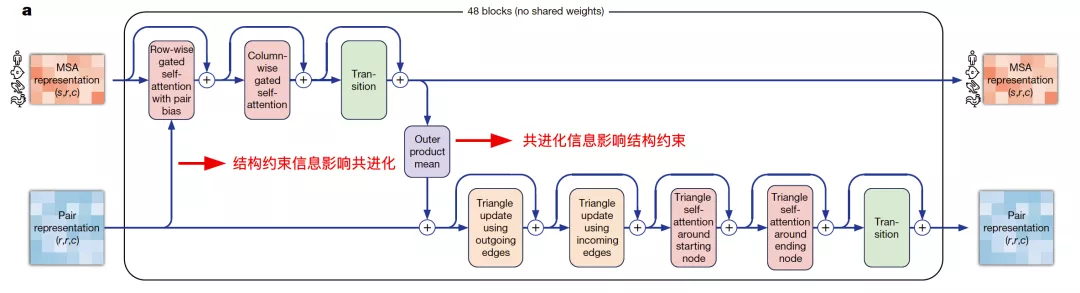
来自:AlphaFold2论文
Structure Module
Structure Module承担着把Evoformer得到的表征解码成蛋白质中每个重原子(C,N,O,S)坐标的任务。为了简化从神经网络预测值到原子坐标的转换,AlphaFold2结合蛋白质中20类氨基酸的结构特性,将重原子分成不同二面角转角决定的组,这样就可以根据给定的起始位置,利用二面角和氨基酸已知的键长键角信息解码出原子坐标。这种结构编码方法相比直接预测坐标(x,y,z)大大降低了神经网络的预测空间,使得端到端结构学习成为可能。

赖氨酸的转角编码方式示例:蓝色平面(C,Cα,Cβ)确定后,根据预测的蓝色-紫色平面的二面角χ1和已知的C-C键长,Cγ-Cβ-N键角即可确定Cγ的空间坐标,重复类似步骤,可以得到Cδ,Cε, N等重原子坐标。
基于飞桨框架的AlphaFold2(AF2)使用
目前已经基于飞桨框架复现了完整的AlphaFold2的inference部分,现已正式在螺旋桨PaddleHelix平台开源:https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/paddlefold
感兴趣的小伙伴们可以安装使用,并基于此,优化自己的蛋白结构预测模型。
1. 安装
在requirements.txt中提供了通过pip可安装的Python依赖项。另外,(基于飞桨框架的AF2还依赖于两个只能通过conda安装的 工具包:openmm==7.5.1和 pdbfixer。为了得到多序列比对MSA,还需要安装kalign, HH-suite 和 jackhmmer。下载数据的脚本需要aria2c。
提供一个可以设置conda环境并安装所有依赖项的脚本setup_env。运行:
sh setup_envconda activate paddlefold # activate the conda environment
也可以在 setup_env中更改环境名称和CUDA版本。
2. 用法
为了运行基于飞桨框架的AF2,还需要蛋白序列数据库和模型参数。基于飞桨框架的AF2使用和AlphaFold2一样的模型参数。
你可以使用脚本scripts/download_all_data.sh来下载和设置所有数据库和模型参数。
运行:scripts/download_all_data.sh <DOWNLOAD_DIR>
将下载完整的数据库。完整数据库的总下载大小约为415 GB,解压后的总大小为2.2 TB。
运行:scripts/download_all_data.sh <DOWNLOAD_DIR> reduced_dbs
将下载一个减少版本的数据库,可以用于在reduced_ dbs的设置下运行。减少的数据库的总下载大小约为190GB,解压缩后的总下载大小约为530GB。
3. 运行基于飞桨框架的AF2进行推理
要使用DeepMind已经训练好的参数对一个序列或多个序列进行推理,运行例如:
fasta_file=\"target.fasta\" # path to the target proteinmodel_name=\"model_1\" # the alphafold model nameDATA_DIR=\"data\" # path to the databasesOUTPUT_DIR=\"paddlefold_output\" # path to save the outputspython3 run_paddlefold.py --fasta_paths=${fasta_file} --data_dir=${DATA_DIR} --small_bfd_database_path=${DATA_DIR}/small_bfd/bfd-first_non_consensus_sequences.fasta --uniref90_database_path=${DATA_DIR}/uniref90/uniref90.fasta --mgnify_database_path=${DATA_DIR}/mgnify/mgy_clusters_2018_12.fa --pdb70_database_path=${DATA_DIR}/pdb70/pdb70 --template_mmcif_dir=${DATA_DIR}/pdb_mmcif/mmcif_files --obsolete_pdbs_path=${DATA_DIR}/pdb_mmcif/obsolete.dat --max_template_date=2020-05-14 --model_names=${model_name} --output_dir=${OUTPUT_DIR} --preset=\'reduced_dbs\' --jackhmmer_binary_path /opt/conda/envs/paddlefold/bin/jackhmmer --hhblits_binary_path /opt/conda/envs/paddlefold/bin/hhblits --hhsearch_binary_path /opt/conda/envs/paddlefold/bin/hhsearch --kalign_binary_path /opt/conda/envs/paddlefold/bin/kalign --random_seed=0
你可以使用python3 run_paddlefold.py -h来查找参数的描述。
保留与AlphaFold2相同的输出,输出将位于output_dir的子文件夹中。它们包括计算的MSAs、模型预测的蛋白结构、OpenMM优化后的结构、模型打分排序、原始模型输出、预测元数据和模型运行计时。output_dir目录将具有以下结构:
<target_name>/ features.pkl ranked_{0,1,2,3,4}.pdb ranking_debug.json relaxed_model_{1,2,3,4,5}.pdb result_model_{1,2,3,4,5}.pkl timings.json unrelaxed_model_{1,2,3,4,5}.pdb msas/ bfd_uniclust_hits.a3m mgnify_hits.stouniref90_hits.sto
每个输出文件的内容如下:
features.pkl
一个 pickle 文件,其中包含模型用于生成结构的输入特性 NumPy 数组。
unrelaxed_model_*.pdb
一个PDB 格式的文本文件,其中包含预测的结构,与模型输出的结构完全一样。
relaxed_model_*.pdb
一个PDB格式的文本文件,是调用OpenMM得到的优化结构,修复了模型预测结构中的冲突,并添加H原子的坐标位置。
ranked_*.pdb
一个 PDB 格式的文本文件,是对OpenMM得到的优化结构按照模型置信度的重新排序。这里使用预测的LDDT分数 (pLDDT)作为置信度评估。
ranking_debug.json
一个JSON格式的文本文件,包含用于执行模型排名的pLDDT值及其对应的模型名称。
timings.json
一个JSON格式的文本文件,包含运行AlphaFold2模型的每个部分所花费的时间。
msas/
该目录中包含不同MSA搜索工具的输出文件。
result_model_*.pkl
一个pickle文件,其中包含一个由模型直接生成的各种 NumPy 数组的字典,除了结构模块的输出外,还包括辅助输出。
最后,可以使用pymol[3]等工具对预测结构和实验结构对齐。值得说明的是,由于输入特征存在采样操作,基于飞桨框架复现的AlphaFold2和JAX版本的预测结构可能会有略微差异,有时候会和实验结构更接近,也可能差别稍大。
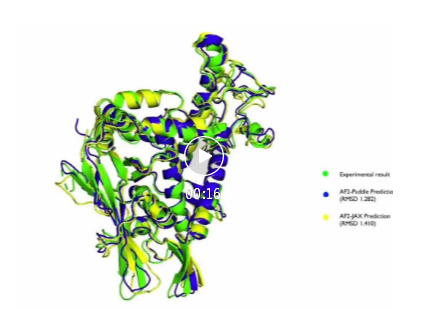
近期开发计划
AlphaFold2虽然在单体蛋白上表现优异,但对复合体,预测的准确度还有待提升。为此,DeepMind团队上线了AlphaFold-Multimer模型,一款针对复合物进行重新训练的神经网络模型,希望能发动飞桨社区开发者们的积极性,一起开发优化基于AlphaFold-Multimer的模型,之后也开源贡献到飞桨平台,让更广大的生信领域研究者们使用基于飞桨框架完全自主可控的蛋白结构预测模型。
本文来源于飞桨PaddlePaddle
来源:http://www.techweb.com.cn/cloud/2022-02-14/2878172.shtml
图文来源于网络,如有侵权请联系删除。